Biopython und Sphinx¶
Notizen¶
Biopython ist ein Paket, das bequeme Funktionen für den Umgang mit Daten in verschiedenen bioinformatischen Standardformaten bereitstellt. Es bietet Schnittstellen für den Zugriff auf Dateien als auch auf Datenbanken im Internet. In der Canopy Python Distribution ist Biopython bereits enthalten. Einen guten Einstieg mit Beispielen finden Sie im Tutorial.
ein Beispielskript für das Einlesen von Sequenzdaten aus einer Datei im Fasta-Format
1from Bio import SeqIO #Import der Funktionen fuer die Ein- und Ausgabe von Sequenzdaten 2 3for seq_record in SeqIO.parse("ls_orchid.fasta", "fasta"): # Einlesen aus einer Datei, der Datensatz landet in der Variable seq_record, die von einem speziellen, durch Biopython definierten Sequenztyp ist 4 print(seq_record.id) # id ist ein spezieller Parameter des Sequenztyps 5 print(repr(seq_record.seq)) 6 print(len(seq_record)) 7
ein Beispielskript, das aus der SwissProt-Datenbank (uniprot_sprot.dat.gz hier als Datei herunterladen - Achtung, sehr groß!) Einträge mit bestimmten Eigenschaften herausfiltert. Das Skript verwendet aus Geschwindigkeitsgründen nicht die o. g. vollständige Datenbank, sondern nur einen kleinen Auszug, der nur die Proteine mit 95-100 Aminosäuren enthält.
1import gzip # Importieren des gzip-Moduls fuer den unkomplizierten Umgang mit gezippten Dateien 2from Bio import SwissProt # Importieren der Funktionen fuer den Umgang mit Daten im SwissProt-Format 3 4 5def PDB_entry(record): 6 """ 7 Extrahiert die PDB-Codes aus den Datensaetzen, die auf vorhandene 3D-Strukturen hinweisen 8 """ 9 PDB_found = [] 10 for entry in record.cross_references: 11 if entry[0] == 'PDB': 12 PDB_found += [entry[1]] 13 return PDB_found 14 15sprothandle = gzip.open(r".\\selected.dat.gz") 16# Oeffnen einer gezippten Datei. Der Zugriff erfolgt dann voellig transparent, d.h. man muss sich nicht selber um das Auspacken kuemmern. 17# Ggf. müssen Sie den Pfad zur Datei anpassen 18 19# sprothandle = open(r".\\selected.dat") 20# Falls Sie das zip-File vorher selber auspacken wollen, öffnen Sie das .dat-File mit diesem Befehl 21 22 23for record in SwissProt.parse(sprothandle): # sequentielles Einlesen der einzelnen Datensaetze 24 25 sequence = str(record.sequence) # Extrahieren der Sequenzinformation fuer den aktuellen Datenbankeintrag 26 27 if sequence.count("C") == 1 and len(sequence) <= 100 and ('3D-structure' in record.keywords): 28 # wenn in der Sequenz nur ein Cystein vorkommt, die Sequenz maximal 100 Aminosaeuren lang ist und auf 3D-Strukturen verwiesen wird 29 a = record.description 30 # print a 31 short_desc = a[a.index("RecName: Full=")+14:].split(';')[0] # Name des Eintrags 32 PDBs = PDB_entry(record) # assoziierte PDB-Codes extrahieren 33 print(record.entry_name, PDBs, short_desc)
ein Beispielskript zur PubMed-Datenbankabfrage über das Netzwerk
1from Bio import Entrez, Medline # Funktionen fuer Entrez und Medline importieren 2 3Entrez.email = "A.N.Other@example.com" # Entrez erwartet eine Nutzeridentifikation 4 5handle = Entrez.esearch(db="pubmed", term="pyruvate oxidase", retmax=3) # Datenbankabfrage von PubMed ueber das Netzwerk. 6 7record = Entrez.read(handle) # Ergebnis wird einem Dictionary-artigen Objekt zugewiesen 8 9# print record, type(record) 10 11idlist = record["IdList"] # Das Feld IdList enthaelt die Datenbankkennungen 12 13# print idlist 14 15ml_handle = Entrez.efetch(db="pubmed", id=idlist, rettype="medline", retmode="text") # Eintraege koennen jetzt aus Pubmed abgerufen werden 16 17ml_records = Medline.parse(ml_handle) 18 19for entry in ml_records: # Ausgabe von verschiedenen Attributen der einzelnen Datenbankeintraege 20 print("title:", entry.get("TI", "?")) 21 print("authors:", entry.get("AU", "?")) 22 print("source:", entry.get("SO", "?")) 23 print()
Sphinx ist ein einfaches System zur Erzeugung statischer Websites, das ursprünglich für die automatisierte Erzeugung der Dokumentation zu Python-Projekten geschrieben wurde. Es ist aber auch generell sehr nützlich. Die Webseite für den Python-Kurs wurde auch damit erstellt. Auch Sphinx ist in Anaconda enthalten bzw. kann mittels conda install sphinx einfach installiert werden. Die Standard-Markup-Sprache, in der die Quelldateien geschrieben werden, heißt RestructuredText. Mit Plugins kann auch Markdown verwendet werden (das z. B. auch für die Wikipedia verwendet wird.). Hier ist der Quelltext für diese Seite als RestructuredText:
Biopython und Sphinx ==================== Notizen ------- - `Biopython <http://www.biopython.org>`_ ist ein Paket, das bequeme Funktionen für den Umgang mit Daten in verschiedenen bioinformatischen Standardformaten bereitstellt. Es bietet Schnittstellen für den Zugriff auf Dateien als auch auf Datenbanken im Internet. In der Canopy Python Distribution ist Biopython bereits enthalten. Einen guten Einstieg mit Beispielen finden Sie im `Tutorial <http://www.biopython.org/DIST/docs/tutorial/Tutorial.html>`_. - ein Beispielskript für das Einlesen von Sequenzdaten aus einer `Datei im Fasta-Format <../../downloads/ls_orchid.fasta>`_ .. literalinclude:: downloads/biopython01.py :language: python :linenos: - ein Beispielskript, das aus der SwissProt-Datenbank (`uniprot_sprot.dat.gz hier <http://ftp.expasy.org/databases/uniprot/current_release/knowledgebase/complete/>`_ als Datei herunterladen - Achtung, **sehr** groß!) Einträge mit bestimmten Eigenschaften herausfiltert. Das Skript verwendet aus Geschwindigkeitsgründen nicht die o. g. vollständige Datenbank, sondern nur einen kleinen `Auszug <../../downloads/selected.dat.gz>`_, der nur die Proteine mit 95-100 Aminosäuren enthält. .. literalinclude:: downloads/biopython02.py :language: python :linenos: - ein Beispielskript zur PubMed-Datenbankabfrage über das Netzwerk .. literalinclude:: downloads/biopython03.py :language: python :linenos: - `Sphinx <http://www.sphinx-doc.org/>`_ ist ein einfaches System zur Erzeugung statischer Websites, das ursprünglich für die automatisierte Erzeugung der Dokumentation zu Python-Projekten geschrieben wurde. Es ist aber auch generell sehr nützlich. Die Webseite für den Python-Kurs wurde auch damit erstellt. Auch Sphinx ist in Anaconda enthalten bzw. kann mittels `conda install sphinx` einfach installiert werden. Die Standard-Markup-Sprache, in der die Quelldateien geschrieben werden, heißt RestructuredText. Mit Plugins kann auch Markdown verwendet werden (das z. B. auch für die Wikipedia verwendet wird.). Hier ist der Quelltext für diese Seite als RestructuredText: .. literalinclude:: VL14_Biopython_Sphinx.rst :language: restructuredtext Auch mathematischer Formelsatz ist einfach möglich: :math:`x_{1,2} = -\frac{p}{2}\pm\sqrt{\left(\frac{p}{2}\right)^2-q}` Aufgaben bis zum nächsten Mal ----------------------------- *keine*
Auch mathematischer Formelsatz ist einfach möglich:
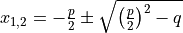
Aufgaben bis zum nächsten Mal¶
keine